The assumption is that the more alike organisms are, the more closely
related they are.
If every "state" is recorded and those states are statistically compared by any method that groups soley on the numbers of states shared between organisms, then there is an inherent assumption of equal rates of evolution because every state is treated as every other and thus must be evolving equally to be compared equally in any equation. This is both the strength and the inherent weakness of simple molecular clock algorithms as well.
Physical pairwise distance comparison techniques, the most famous of which was DNA hybridization are historical now.
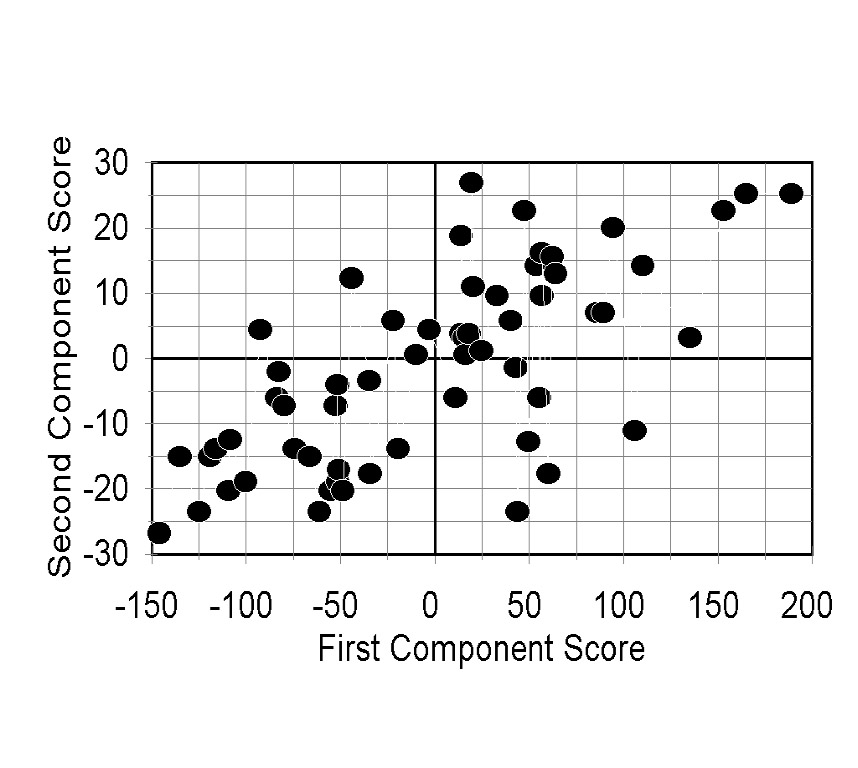
PCA
Principal Components Analyses
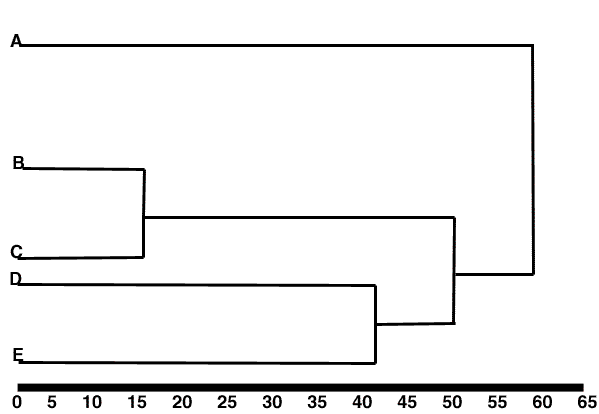
UPGMA/WPGMA
(un)weighted pair group method using arithmetic averages
These trees are fast to compute and provide a useful representation of the data (i.e. they might be useful in telling you if you need to sequence more in a big molecular study) but they are not useful for generating phylogenies for the reasons discussed above.
Here is a link to a very brief example by Steven Carr.